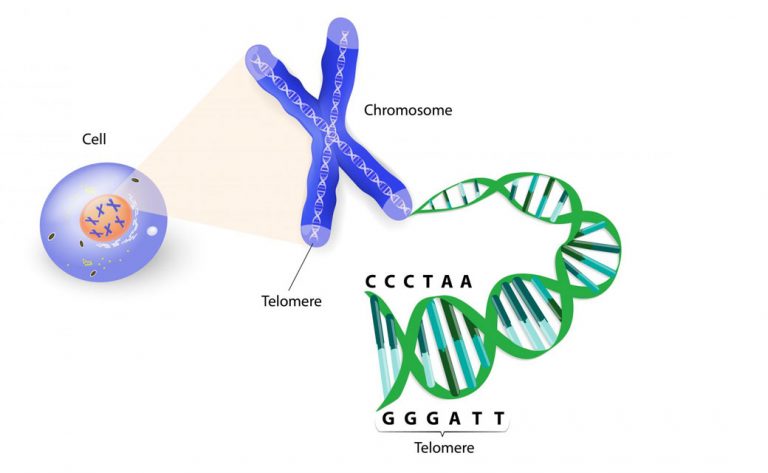
The length of telomeres, the chromosomes’ end tails, as depicted in the figure below, physiologically shorten with aging. This means that younger cells have longer telomeres compared to their aged counterparts; one could say that the length of cells’ telomeres is a measure of their replicative power. Cancer cells are known to be able to turn this into their favour, and expand their replicative immortality through telomere lengthening and maintenance. Despite being a well-studied pan-cancer hallmark, the relation of the telomeres length to specific cancer genomic features is poorly understood.
Julia Livingstone and her colleagues from P.C. Boutros' team in Los Angeles focused their work on prostate cancer. To investigate the relations between telomere length and the clinico-genomics of prostate tumours, they used whole genome sequencing (WGS) of 392 published tumour–reference pairs previously deposited at the EGA under accession numbers EGAS00001000400 and EGAS00001000900. Their work was published on Nature Communications in November 2021.
They determined the telomere length of the sequencing sample, from both tumour and non-tumour adjacent tissues, and subsequently ran statistical analysis to investigate possible correlation between the telomeres length with several cancer features. The team discovered that telomeres length does not correlate with any specific driver gene mutation, but it does with the overall frequency of single nucleotides variants in a sample. This finding suggests that tumours with shorter telomeres accumulate more mutations without strong selective pressures for specific ones. While proliferation rate did not correlate directly with telomere length, methylation of almost half of all genes strongly did. The team’s results are massive and reveal a complicated and fascinating relation of different clinical and genomic features of prostate cancer with telomere length. In the paper discussion, they smartly consider different possibilities.
As noted above, all these amazing results have been obtained analysing previously sequenced samples. Remarkably, the researchers who generated the samples did not focus at all on telomeres length. They indeed studied specific genes mutations and the features of early and late-onset prostate cancer. This proves, once again, the power of sequencing data re-use. This data is tremendously rich in information, and can hardly be exhausted with any number of analyses. Generating sequencing data, especially Whole Genome Sequencing, even better if tumor-reference paired, is like shopping every last article at the grocery store. You can then cook several recipes, or prepare a seven-course dinner, and still have ingredients left for someone else to borrow and cook something you have not thought about, a recipe that is just not in your cookbook.
Noteworthy, the data retrieved from the EGA for this paper is associated with rich and complete metadata, meaning pheno-clinical information regarding the sample donors. As Julia Livingstone and her team wrote, using rich clinical annotations enabled them to assess the relationship between telomere length and patients outcome. This highlights once again the importance of reporting and annotating metadata correctly; It might feel like extra work for the researchers depositing their data, but it really has a huge value. Our team at the EGA is currently working to improve the metadata annotation process, with the aim to maximize the benefits for the scientific community.